Tay-Sachs Disease: Delve into Diseases & Disorders
- Lasya Kambhampati
- Oct 18, 2020
- 4 min read

Tay-Sachs disease (also known as Type 1 GM2 Gangliosidosis) is a rare genetic condition that destroys the cells of the central nervous system. Loss of the enzyme that breaks down gangliosides, fatty substances, is the root cause. It is often seen in infants and has a short life expectancy as there is no cure for the deadly disease. Today we will be exploring the genetics, symptoms, specific risk groups, diagnosis, and potential treatments.
Genetics
The genetic basis of Tay-Sachs is a mutation in the gene that encodes the Hex-A enzyme or hexosaminidase. Hex-A plays a large role in the central nervous system (CNS) and can be found in lysosomes. The enzyme, as stated above, is essential in degrading gangliosides. The gene is found on the long arm (q) of chromosome 15. There are over 40 possible mutations that can cause the gene to stop functioning. Since Tay-Sachs is an autosomal recessive disease, patients need to have inherited two copies of a mutation gene. This means that both parents must be carriers for this mutation (heterozygotes).


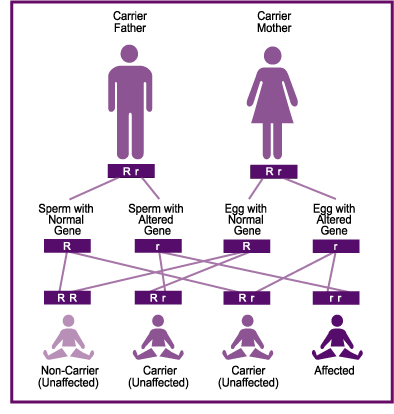
Because Tay-Sachs requires two copies of the gene and is relatively rare, it is not a major concern for most parents. However, there are certain groups that have a significantly higher risk than the rest of the population. These groups are Ashkenazi Jews, some French Canadian communities in Quebec, an Old Order Amish community in Pennsylvania, and the Cajun community of Louisiana. Testing for Tay-Sachs can be crucial for members of these high-risk groups.
Parents and children can be easily tested for Tay-Sachs. A quick blood test can determine whether or not parents carry the mutation. This can help inform their decision to have a child and encourage them to see a genetic counselor. Tay-Sachs can also be diagnosed during the pregnancy (prenatally) through amniocentesis or chorionic villus sampling (CVS). Amniocentesis samples the fluid around the fetus and CVS uses a tissue sample from the placenta. The samples can be tested for the disease through an enzyme assay or if the specific mutation is known, a molecular genetic test can be used.
Symptoms
Tay-Sachs is most commonly diagnosed when the patient is an infant. Although extremely rare, Tay-Sachs can also occur later in life. These patients can have milder symptoms depending on the time of onset.

Infantile Tay-Sachs
The symptoms can include loss of motor skills or weak muscles, adverse reactions to loud noises, seizures, partial or complete loss of vision or hearing. Patients often also have trouble swallowing or chewing, as a result of their weakened muscles. Red spots in the eye are an extremely important diagnostic factor.
Children often also experience severe respiratory infections, paralysis, or intellectual disabilities. Unfortunately, as there is no cure and Tay-Sachs has many complications, patients only live for a few years (if they have the most common form).
Juvenile Tay-Sachs Disease
This version of Tay-Sachs can happen between the ages of 2-10. Ataxia (issues with voluntary muscle movements) is one of the most common first symptoms. Similar to infantile Tay-Sachs, patients experience a variety of speech issues as well as intellectual disabilities. Although patients may not have the cherry-red spots in their eyes, they can have optic atrophy (degeneration of the nerve connecting the eye to the brain) or retinitis pigmentosa (progressive degeneration of the retina). Although patients have a longer life-span than those with infantile, complications from Tay-Sachs often result in death around the age of 15.
Late-Onset Tay-Sachs Disease
Individuals with late-onset Tay-Sachs often have more varied symptoms. The disease has a much slower progression which may play a role in the variability of the symptoms. Some general symptoms include clumsiness, muscle atrophy, and mood swings. As time goes on, symptoms tend to worsen, especially when combined with the effect of aging. Patients can experience seizures, muscle tremors or twitching, difficulty controlling muscles, and issues swallowing. Individuals can also experience involuntary muscle contractions (dystonia) that can be quite painful. Due to the breadth of muscle issues that patients face, they often experience problems walking or running, or exercising and may even require braces or a wheelchair. There are also quite a few mental effects: deterioration of concentration, issues with memory, difficulty concentrating, and personality changes. Furthermore, around 40% of patients experience bipolar episodes, hallucinations, paranoia, or depression.
Treatments
Although there is no cure for Tay-Sachs or effective treatments, there are promising research developments that could change that.
Enzyme-replacement therapy
Researchers are currently investigating the plausibility of using enzyme-replacement therapy on patients with Tay-Sachs. Enzyme replacement therapy is exactly what it sounds like; it replenishes a specific enzyme in patients that do not have enough. Synthetic versions are developed and then injected into the individual. The treatment has been shown the be successful in other diseases, even lysosomal storage diseases. However, this treatment has issues for patients with Tay-Sachs. Since Tay-Sachs is primarily a neurological disease, the enzyme needs to cross the blood-brain barrier (see this article to learn more). Researchers are working to overcome this obstacle in order to treat patients.
Gene Therapy
Gene therapy is quite similar to enzyme-replacement therapy but targets the mutated gene instead of the enzyme. The treatment involves replacing the defective gene with a normal copy in order to produce the enzyme in normal quantities. However, there are technical and ethical issues with the gene therapy that have yet to be resolved.
Chaperone therapy
Chaperone therapy involves small molecules binding to the mutated hexosaminidase A enzymes and guiding them to the lysosome preventing their degradation. Although the enzyme is mutated, it may be able to perform some of its original function in the lysosome. Since the chaperone can cross the blood-brain barrier, it does not need to overcome that obstacle but does need further testing to prove its effectiveness.
Similar Diseases: Sandhoff Disease
Because Tay-Sachs requires two copies of the gene and is relatively rare, it is not a major concern for most parents. However, certain groups have a significantly higher risk than the rest of the population. These groups are Ashkenazi Jews, some French Canadian communities in Quebec, an Old Order Amish community in Pennsylvania, and the Cajun community of Louisiana. Testing for Tay-Sachs can be crucial for members of these high-risk groups. tor issues, and intellectual impairment progress with time. Patients may have sensory loss or seizures. Although Sandhoff is caused by mutations in the HEXA and HEXB genes and is inherited autosomal recessively, it does not have high-risk groups and can found in equal rates in the general population.



Comments